The Real Reason VO₂ Max Declines With Age — And Why It Changes How You Should Train
Prefer to listen? Hit play for a conversational, audio‑style summary of this article’s key points.
VO₂ max is one of the strongest predictors of longevity, and it declines relentlessly. Beginning around age 30, VO₂ max drops at roughly 10% per decade in sedentary individuals, a trajectory that can eventually fall below the threshold required for basic daily function.
The decline is not just about the heart. VO₂ max falls by approximately 46% between ages 20 and 70, while maximal cardiac output falls by only 31% over the same period. The gap between those two numbers is the first quantitative signal that something beyond cardiac output is driving the loss.
By late middle age, nearly half of the limitation on VO₂ max is peripheral in origin. In younger adults, approximately 77% of the total limitation is central and 23% peripheral. In older adults, that splits to roughly 56% central and 44% peripheral. The muscles are catching up to the heart as a source of failure.
Oxygen extraction drops significantly with age. Skeletal muscle extracts roughly 80% of delivered oxygen at maximal effort in young adults. By ages 75 to 80, that figure falls to approximately 60%, a 20 percentage point decline in a variable that most aging research has historically underemphasized.
The peripheral decline reflects four converging biological processes. Sarcopenia preferentially strips away mitochondria-rich type II muscle fibers. Mitochondrial density and efficiency decline. Capillary networks thin, increasing diffusion distances. And interstitial changes further impair oxygen movement from blood to cell. None of these is catastrophic alone, but together they compound into something substantial.
The oxygen cascade is trainable. The biological machinery driving peripheral decline, mitochondria, capillaries, and oxidative enzymes, remains responsive to exercise well into the later decades of life. The system that aging erodes is the same system that training can partially rebuild.
Endurance training is the most reliable builder of capillary networks. Over 8 to 10 weeks, it produces a 13.3% increase in capillary density and a 15% increase in capillary-to-fiber ratio, the strongest and most consistent vascular gains of any modality. It is the foundation of long-term peripheral oxygen extraction capacity.
HIIT is the most time-efficient route to mitochondrial adaptation. It produces comparable mitochondrial gains to endurance training while requiring substantially less total training time, estimated at approximately 1.7 times more time-efficient for driving mitochondrial remodeling. For most people balancing real life with long-term healthspan, it represents the highest-yield option.
SIT delivers the fastest mitochondrial signal per minute of any modality. When gains are normalized to total training time, SIT generates three to five times greater VO₂ max improvement per hour of exercise than either endurance training or HIIT. Its tradeoff is vascular remodeling: capillary density shows no significant average increase with SIT alone, making it a powerful complement to endurance training rather than a replacement for it.
Meaningful adaptation begins faster than most people assume. Approximately 13.7% of total mitochondrial gains from an endurance training block occur within the first two weeks. Capillary remodeling begins similarly early, though it tends to plateau around four weeks without progressive increases in volume or intensity.
The decisions made in the fourth and fifth decades of life shape the physiological ceiling of the seventh and eighth. Aerobic aging is a slow, cumulative process across multiple systems simultaneously. So is the adaptive response to training. The most important variable is not the perfect modality. It is consistency across the decades during which the oxygen cascade is quietly remodeling in one direction or the other.
Introduction
There are few physiological markers that decline with age as reliably or as consequentially as maximal aerobic capacity, better known as VO₂ max.
VO₂ max represents the ceiling of the body's oxygen engine: the highest rate at which a person can take in, transport, and utilize oxygen during all-out effort. It sounds like an athlete's metric, and it is, but it's also something more fundamental. Across decades of epidemiological research, VO₂ max has emerged as one of the most powerful predictors of longevity, cardiovascular health, and the ability to live independently as we age. In some analyses, low cardiorespiratory fitness rivals smoking as a risk factor for premature death. When VO₂ max falls, our functional capacity tends to fall with it.
The decline is relentless. Beginning around the fourth decade of life, VO₂ max drops at roughly 10% per decade in sedentary individuals, a trajectory that, left unchecked, can eventually cross below the threshold required for basic daily function. Exercise slows that decline considerably, but does not stop it.
For over a century, exercise physiologists haven't debated whether VO₂ max declines. That much is indisputable. The real question has been why.
What, precisely, limits aerobic capacity as we grow older?
The answer lies in a deceptively elegant physiological equation. At its core, VO₂ max is governed by what's known as the Fick principle, which states that the body's total oxygen consumption is the product of two things: how much blood the heart can pump per minute (cardiac output) and how much oxygen the muscles can extract from that blood, known as the arteriovenous oxygen difference. In equation form:
VO₂ = Cardiac Output × Arteriovenous Oxygen Difference
This single equation divides the mystery of aerobic aging into two distinct physiological stories.
The first is a story about the heart. Cardiac output is itself the product of two variables: heart rate and stroke volume, meaning the amount of blood pumped with each beat. Both decline with age. Maximal heart rate falls predictably, at roughly one beat per minute per year after age 20. Stroke volume can also decrease as the heart muscle stiffens with age, the large arteries lose elasticity, and the ventricles fill less completely with each cycle. Together, these changes reduce the total volume of oxygen-rich blood the body can deliver to its working muscles. Historically, this "central" limitation has been treated as the dominant explanation for why VO₂ max declines with age.
But delivering oxygen is only half the story.
The second story is about what happens once that blood arrives at muscle tissue. The muscles must actually extract and use the oxygen, and that capacity depends on an entirely different set of biological structures: the density of capillaries threading through muscle tissue, the number and health of mitochondria (the cellular organelles that convert oxygen into usable energy), the mass of the muscle itself, and the activity of the enzymes that drive oxidative metabolism. These are the "peripheral" factors, and aging affects all of them. Muscle mass declines through a process called sarcopenia. Capillary networks thin out, a phenomenon called capillary rarefaction. Mitochondria become fewer in number, smaller in size, and less efficient at producing energy. Oxidative enzyme activity wanes.
Each of these changes could theoretically reduce the amount of oxygen muscles pull from the bloodstream. But historically, their collective contribution has been harder to pin down. Cardiac output can be estimated relatively straightforwardly through imaging and hemodynamic measurements. Mitochondrial density and peripheral oxygen extraction, by contrast, require more invasive and technically demanding methods, including biopsies, sophisticated metabolic probes, and near-infrared spectroscopy, that are difficult to deploy across large study populations. As a result, the peripheral side of the Fick equation has received far less systematic attention, and a seductively simple narrative has dominated the field: VO₂ max declines because the aging heart pumps less blood.
But what if that explanation is incomplete?
A new study takes aim at this gap, providing more precise quantification than previous work of how much the central and peripheral components each contribute to the age-related decline in maximal aerobic capacity [1]. The findings complicate the old story in important ways. Aging, the data suggest, is not simply a tale of a slowing heart, but of coordinated remodeling across the entire oxygen transport chain, from the cardiac muscle to the smallest capillaries to the mitochondria embedded within skeletal muscle fibers.
That distinction matters enormously, not just for how we understand the biology of aging, but for how we train, how we intervene, and how we think about preserving physiological resilience across a lifetime.
How Do You Measure What's Limiting VO₂ Max?
To answer the central question of what really drives the age-related decline in VO₂ max, the researchers needed a way to separate delivery from utilization. That sounds straightforward. In practice, it's anything but.
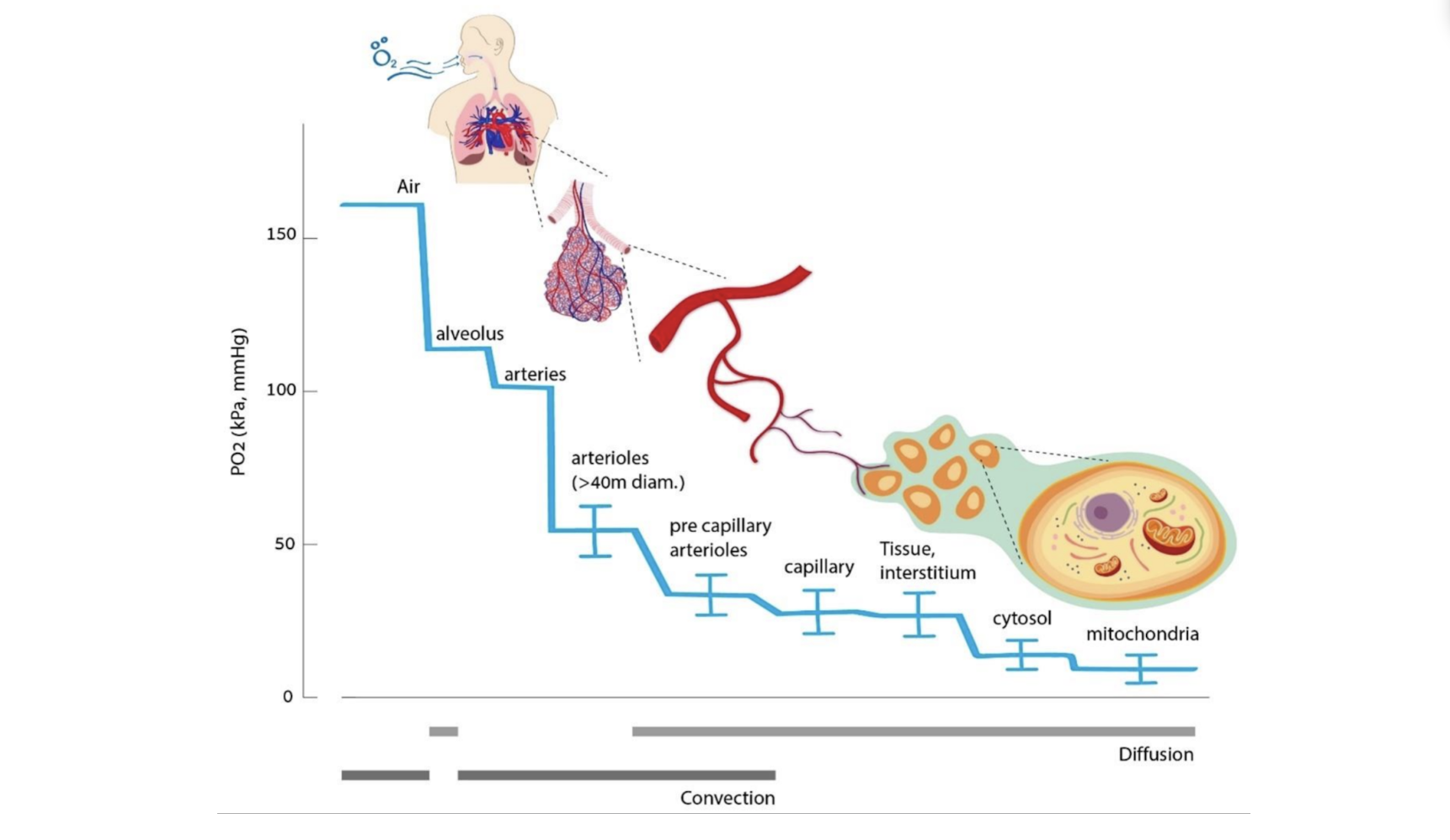
Oxygen doesn't simply arrive in muscle tissue the way water comes out of a tap. It travels through what physiologists call the oxygen cascade, a stepwise journey from the atmosphere to the interior of individual cells. With each step, the path narrows, and the environment changes. Oxygen moves from air into the lungs, dissolves across the thin membranes of the alveoli into the bloodstream, gets bound to hemoglobin inside red blood cells, and is then carried through the heart and into the arterial system. From there, it passes through progressively smaller vessels, crosses capillary walls, diffuses through the fluid-filled space between cells, enters muscle fibers, and finally reaches the mitochondria, where it is consumed in the process of making ATP, the molecular currency of cellular energy.
At every one of these steps, there is some resistance to flow. The oxygen cascade is less like a pipe and more like a series of filters, each one imposing its own constraints on how much oxygen can move through.
This is where the study's central methodological insight comes in. Rather than trying to measure each individual step of the cascade separately, which would be technically forbidding across large populations of people spanning different ages, the researchers borrowed a framework from an unlikely source: electrical engineering.
In a circuit, when electrical current passes through multiple resistors arranged in sequence, each resistor imposes its own drag on the flow of current. The total resistance of the system is simply the sum of the individual resistances. The researchers applied this same logic to the oxygen cascade, treating the biological barriers that oxygen must cross not as a tangle of separate physiological processes but as a system of resistors in series. The "current" in this analogy is the flow of oxygen through the body. The resistors are the biological structures that slow it down.
By modeling the system this way, they could collapse the cascade into two composite quantities that map cleanly onto the two sides of the Fick equation introduced earlier.
Cardiovascular Resistance (RQ)
The first is cardiovascular resistance, which the researchers labeled RQ. This captures everything involved in oxygen delivery: cardiac output, the flow of blood through the large arteries, and the broader dynamics of systemic circulation. In essence, RQ reflects the question: how powerfully can the heart and vasculature push oxygenated blood toward working muscles?
Peripheral Resistance (RP)
The second is peripheral resistance, labeled RP. This captures everything that happens once oxygen reaches the smallest vessels and beyond: the density of capillaries within muscle tissue, the rate at which oxygen diffuses across capillary walls and into muscle fibers, and the capacity of mitochondria to consume it. RP reflects the question: once oxygen arrives at the muscle, how effectively can the tissue actually use it?
Together, RQ and RP account for the full journey of oxygen from heart to mitochondrion. And by estimating how each of these resistances changes with age, the researchers could, for the first time with this level of precision, assign quantitative responsibility for the age-related decline in VO₂ max to each side of the system.
Central vs. Peripheral Limitation
With the resistance framework in place, the researchers could now ask a deceptively simple question: for any given level of VO₂ max, how much of the limitation comes from the cardiovascular system, and how much comes from the periphery?
They quantified this by calculating what they called the fractional central limitation and the fractional peripheral limitation. Think of these as percentages of blame. If the cardiovascular system is responsible for 60% of the gap between a person's actual VO₂ max and its theoretical ceiling, and the peripheral tissues account for the remaining 40%, those fractions tell you where the real bottleneck lies. And critically, as a person ages, those fractions can shift, even if both systems are declining simultaneously.
A key variable in this analysis was maximal cardiovascular oxygen delivery: the total volume of oxygen the heart and vasculature can deliver to the body's tissues during all-out exercise. This is essentially the output of the central system, the raw supply of oxygen available to the muscles at peak effort.
But supply alone doesn't determine performance. A fire hydrant is useless if the pipes leading to it are clogged. Even when the cardiovascular system delivers a generous volume of oxygen, that oxygen only contributes to VO₂ max if the muscles can actually extract and consume it. So the researchers also examined what fraction of delivered oxygen the peripheral tissues actually pull out of the bloodstream, a ratio that serves as a direct index of peripheral effectiveness.
When that extraction fraction is high, the peripheral system is doing its job well: capillaries are dense, diffusion is efficient, and mitochondria are plentiful and active. When it is low, something downstream is failing to keep up. The oxygen arrives but goes underutilized, like food on a plate that never gets eaten.
Where Did the Data Come From?
Rather than recruiting subjects and running new experiments, the researchers took a different and arguably more powerful approach. They compiled existing published data drawn from decades of exercise physiology research, assembling measurements of VO₂ max and maximal cardiac output from mostly healthy, physically active men between the ages of 30 and 90.
This kind of synthesis has real advantages. No single laboratory study can easily recruit participants spanning six decades of age under controlled conditions. But the accumulated literature, when carefully curated, contains exactly that breadth. By pooling data across studies, the researchers could examine how VO₂ max and cardiac output change across the adult lifespan with a statistical power that no individual experiment could match.
Because both VO₂ max and cardiac output were available in the dataset, the researchers could estimate maximal oxygen delivery directly from the data. From there, their resistance model allowed them to mathematically partition the total limitation into its central and peripheral components, essentially reconstructing the bottleneck without ever having to measure peripheral oxygen extraction directly.
What emerged from that reconstruction challenges one of the most entrenched assumptions in the field. The age-related decline in VO₂ max, it turns out, is not primarily a story of a weakening heart.
VO₂ Max and Cardiac Output Decline—Just Not Equally
When the researchers plotted maximal aerobic capacity across adulthood, the pattern was exactly what any exercise physiology textbook would predict: both VO₂ max and maximal cardiac output declined steadily with age. But they did not decline at the same rate, and that asymmetry turns out to be enormously revealing.
On average, VO₂ max fell by approximately 37 milliliters of oxygen per minute per year. Maximal cardiac output fell by roughly 151 milliliters of blood per minute per year. Translated across the span from age 20 to 70, these rates compound into something striking: a 46% reduction in VO₂ max against only a 31% reduction in maximal cardiac output.
Aerobic capacity, in other words, falls substantially faster than the heart's pumping capacity alone would predict. That gap is not a rounding error or a statistical artifact. It is a signal.
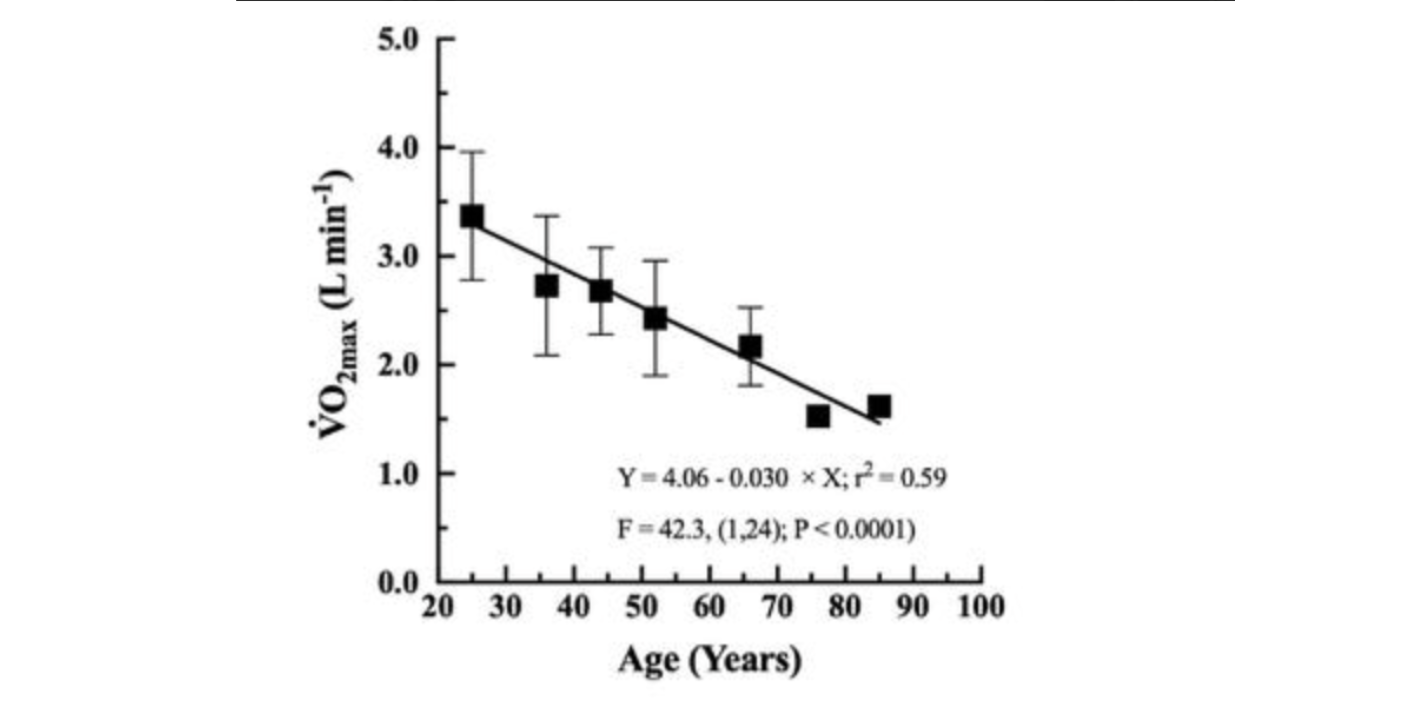
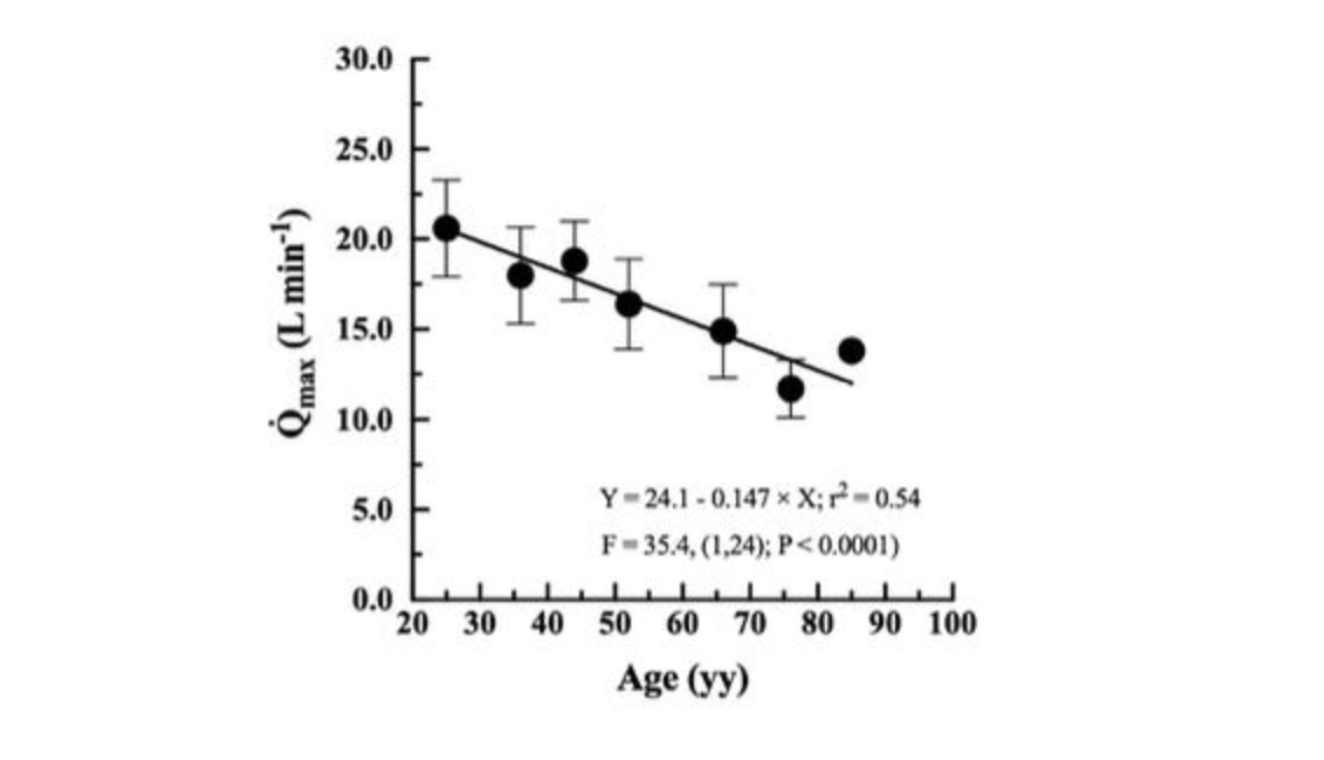
Here is why it matters. The Fick equation, introduced earlier, tells us that VO₂ max is the product of cardiac output and the fraction of oxygen extracted by the tissues. If cardiac output and VO₂ max declined at the same proportional rate, we could reasonably conclude that the heart is the primary driver of the loss, and that peripheral oxygen extraction is holding roughly steady with age. But that is not what the data show. VO₂ max is falling faster than cardiac output, which means the oxygen extraction side of the equation must also be deteriorating. The tissues are not just receiving less oxygen; they appear to be doing less with what they receive.
From a biological standpoint, this makes sense. The peripheral machinery for oxygen utilization is not static with age. Capillary networks thin through a process of rarefaction, increasing the average distance between a blood vessel and the muscle fibers it supplies. Skeletal muscle fibers atrophy and are partially replaced by connective tissue and fat, reducing the metabolically active mass available to consume oxygen. Mitochondria, which carry out the final step of the oxygen cascade by using it to drive the production of ATP through a process called oxidative phosphorylation, become fewer in number and less efficient with age. The molecular assembly lines inside those mitochondria, the enzyme complexes of the electron transport chain, also decline in activity.
The cumulative effect is a peripheral system that receives oxygen but captures less of it. The supply chain is impaired, but so is the factory at the end of it.
The unequal rates of decline in VO₂ max and cardiac output provide the first quantitative fingerprint of this dual deterioration. And what the resistance modeling allowed the researchers to do next was move beyond this first clue and directly calculate how much of the total age-related loss in VO₂ max could be assigned to each side of the system.
VO₂ max is falling faster than cardiac output, which means the oxygen extraction side of the equation must also be deteriorating. The tissues are not just receiving less oxygen; they appear to be doing less with what they receive.
With Age, Muscles Use Less of What the Heart Delivers
The most revealing finding emerged when the researchers examined maximal oxygen extraction: the fraction of delivered oxygen that working muscle tissue actually pulls from the bloodstream during peak effort. This single variable, sitting quietly on the peripheral side of the Fick equation, tells a story that the cardiac output data alone cannot.
In young adults around age 20, skeletal muscle extracts roughly 80% of the oxygen delivered to it at maximal effort. By ages 75 to 80, that figure has fallen to approximately 60%. A 20 percentage point drop across the adult lifespan, in a variable that most aging research has historically underemphasized.
To appreciate why this matters, consider what extraction efficiency actually represents at the cellular level. When oxygen-laden blood arrives at a capillary bed within muscle tissue, the rate at which oxygen moves from the bloodstream into the muscle fiber depends on several factors: the density of capillaries (which determines how much surface area is available for exchange), the distance oxygen must diffuse to reach a mitochondrion, and the metabolic demand of the mitochondria themselves. If capillaries have thinned, if muscle fibers have atrophied and the geometry of the tissue has changed, or if the mitochondria have lost mass and enzymatic capacity, less oxygen gets pulled across the capillary wall per unit of blood that flows past. The extraction fraction falls. In the language of the resistance model, peripheral resistance has risen.
And when the researchers quantified how that rising peripheral resistance reshapes the balance of limitation across the lifespan, the results were striking.
In younger adults, approximately 77% of the total limitation on VO₂ max is attributable to the central cardiovascular system, with only 23% attributable to the periphery. The heart and major vessels are the dominant bottleneck. The muscles, capillaries, and mitochondria are largely keeping pace with whatever oxygen the cardiovascular system delivers.
But that balance shifts substantially with age. In older adults, the central contribution to limitation drops to around 56%, while the peripheral contribution rises to approximately 44%. The two sides of the oxygen transport system are converging toward parity.
What this means in practical terms is that the peripheral machinery, the microvasculature threading through muscle, the mitochondria embedded within individual fibers, and the enzymes driving oxidative metabolism, becomes nearly as limiting to aerobic capacity as the heart itself. The muscles, in a sense, catch up as a source of failure. The cardiovascular system is still declining, but the tissues are declining alongside it, and increasingly on their own terms.
This represents a meaningful shift in how we should conceptualize aerobic aging. The dominant narrative, that VO₂ max falls because the aging heart pumps less blood, is not wrong. But it is increasingly incomplete as a person grows older. By the seventh and eighth decades of life, roughly half of the total limitation on maximal aerobic capacity is peripheral in origin. The problem is not just supply. It is also, and perhaps equally, the capacity to use what is supplied.
The model further suggests that this progressive rise in peripheral resistance may be at least as important as the decline in maximal cardiovascular oxygen delivery in driving the overall trajectory of aerobic aging. That is a finding with direct implications for how we think about intervention, a point we will return to.
By the seventh and eighth decades of life, roughly half of the total limitation on maximal aerobic capacity is peripheral in origin. The problem is not just supply. It is also, and perhaps equally, the capacity to use what is supplied.
Why Does Oxygen Extraction Decline?
The resistance model used in this study is a mathematical framework, not a microscope. It cannot directly observe what is happening inside aging muscle tissue. But its findings align closely with decades of cellular and molecular research on skeletal muscle aging, and the biological mechanisms that likely drive the rise in peripheral resistance are well characterized.
Several converging processes appear to be responsible.
- Sarcopenia
The first is sarcopenia, the age-related loss of skeletal muscle mass and strength that begins in the fourth decade of life and accelerates thereafter. Sarcopenia is not simply a matter of muscles getting smaller. It preferentially strips away type II muscle fibers, the fast-twitch fibers that are particularly rich in mitochondria and oxidative capacity, while leaving the slower, less metabolically active type I fibers relatively intact. The result is a muscle that is not only smaller but compositionally different, with a reduced density of the very organelles responsible for consuming oxygen.
- Mitochondrial Loss and Dysfunction
The second process unfolds inside those remaining mitochondria. Aging muscle shows a well-documented decline in mitochondrial density, meaning there are simply fewer of these organelles per unit of muscle volume. But quantity is not the only problem. The mitochondria that remain also function less efficiently. The protein complexes of the electron transport chain, the molecular machinery that uses oxygen to generate ATP, decline in activity. Mitochondria in older muscle also produce more reactive oxygen species, unstable molecules that can damage cellular components including the mitochondria themselves, creating a self-reinforcing cycle of dysfunction. The net result is that even when oxygen successfully crosses the capillary wall and enters a muscle fiber, the cell's capacity to convert it into usable energy may be substantially reduced.
- Capillary Rarefaction and Microvascular Dysfunction
The third process operates at the level of the microvascular network. Capillary density within skeletal muscle can decline with age, a phenomenon that carries significant geometric consequences. Each capillary supplies oxygen to the muscle fibers in its immediate vicinity, so when capillaries are lost, the average distance that oxygen must diffuse to reach a mitochondrion increases. Because diffusion is a slow process that depends on concentration gradients, even modest increases in diffusion distance can meaningfully reduce the rate at which oxygen moves from blood into tissue. Compounding this, the endothelial cells lining the blood vessels become less responsive with age, impairing the vasodilation that normally routes blood toward the most metabolically active regions of muscle during exercise.
- Diffusion Limitations
The fourth process is more subtle but operates along the same diffusion pathway. Changes in the composition of the interstitial space, the fluid-filled gap between capillaries and muscle fibers, as well as shifts in muscle fiber geometry as tissue is partially replaced by fat and connective tissue, can increase the structural resistance that oxygen must overcome on its way from the bloodstream to the mitochondrion.
Taken individually, none of these changes is catastrophic. A modest loss of capillary density, a modest reduction in mitochondrial efficiency, a modest increase in diffusion distance. But they do not occur in isolation. They accumulate simultaneously, in the same tissue, over the same decades, and their effects on peripheral oxygen extraction compound one another. The oxygen cascade becomes progressively less permeable at every step downstream of the capillary wall.
The cumulative result is a peripheral system that extracts less oxygen per unit of blood delivered, not because any single mechanism has failed dramatically, but because many mechanisms have each failed a little, and together they add up to something substantial.
Understanding why extraction declines, however, also reveals something more hopeful: most of the biological machinery responsible is not fixed. It is responsive. And exercise is one of the most potent stimuli known to remodel it.
The Oxygen Cascade Is a Trainable System
The modeling framework at the heart of this study does something subtle but important. It reframes VO₂ max not as a fixed number stamped into a person's biology, but as the output of a system, one built from interconnected components that each contribute to the final result. And systems, unlike simple numbers, can remodel.
This is not merely an academic distinction. If the age-related decline in VO₂ max were driven entirely by the heart, the therapeutic implications would be relatively narrow. Cardiac output is not easily modified by lifestyle once aging has taken hold. But if peripheral resistance is contributing nearly as much as central limitation by late middle age, and if that peripheral resistance reflects the state of mitochondria, capillaries, and diffusion geometry within skeletal muscle, then the picture changes considerably. These are structures that respond to training. The question is how. In the sections below, we will structure training to target these components of the peripheral system.
Mitochondrial Remodeling: The Decline in Oxygen Utilization
If younger muscle extracts roughly 80% of delivered oxygen and older muscle extracts closer to 60%, the obvious question is: what has changed inside the cell?
The most well-documented answer involves mitochondria, and it operates on two levels simultaneously: how many there are, and how well they work.
On the quantity side, multiple biopsy studies have confirmed that older skeletal muscle contains fewer mitochondria per unit of tissue volume. This reduction traces back partly to a decline in mitochondrial biogenesis, the cellular process by which new mitochondria are generated. In younger muscle, repeated bouts of intense exercise create metabolic stress that activates a master regulatory protein called PGC-1α, which in turn coordinates a broad transcriptional program: genes are switched on, mitochondrial proteins are synthesized, and the mitochondrial network expands. This is one of the primary biological mechanisms by which exercise improves aerobic fitness. With aging, however, the signaling cascade that links exercise stress to PGC-1α activation can become blunted. The same stimulus that once triggered robust mitochondrial expansion produces a smaller molecular response. Fewer new mitochondria are built, and the ones that are lost to normal cellular turnover are not fully replaced.
On the quality side, the mitochondria that remain in older muscle are themselves less capable. The protein complexes of the electron transport chain, the series of molecular machines embedded in the inner mitochondrial membrane that use oxygen to drive ATP synthesis, decline in both number and efficiency. Older mitochondria also tend to leak protons across their inner membrane, a process that dissipates the electrochemical gradient that powers ATP production and generates heat instead of energy. They produce more reactive oxygen species, chemically unstable molecules that can damage proteins, lipids, and DNA, including within the mitochondria themselves, creating a degenerative feedback loop. The balance between mitochondrial fission (splitting into smaller units) and fusion (merging into larger networks) is also disrupted with age, and the cellular quality-control process called mitophagy, which normally clears damaged mitochondria before they can cause harm, becomes less efficient. The result is an accumulation of dysfunctional organelles that consume resources without contributing meaningfully to aerobic metabolism.
Fewer mitochondria, each operating below capacity. The extraction fraction falls.
Capillaries and Microvascular Aging: The Hidden Bottleneck
If mitochondria are the engines of aerobic metabolism, capillaries are the roads that bring them fuel.
Oxygen does not leap directly from artery to cell. After leaving the heart, it travels through a branching network of progressively smaller vessels, from large arteries to arterioles, before arriving at the capillaries, the finest vessels in the body, often just wide enough for a single red blood cell to pass through in single file. It is here, at this microscopic interface, that oxygen actually crosses from the bloodstream into tissue. It diffuses across the thin endothelial wall of the capillary, through a narrow interstitial space, into the muscle fiber, and finally to the mitochondrion waiting to consume it. That final stretch, measured in microns, may be one of the most consequential determinants of peripheral oxygen extraction.
With aging, the microvascular network remodels in ways that progressively impair this final step. Capillary density declines, meaning fewer capillaries per cross-sectional area of muscle. Because each capillary supplies a surrounding halo of tissue, losing capillaries increases the average distance oxygen must diffuse to reach a mitochondrion. Simultaneously, the endothelial cells lining these vessels become less responsive to the chemical signals that trigger vasodilation during exercise. In particular, production of nitric oxide, a gaseous signaling molecule that causes smooth muscle in vessel walls to relax and the vessel to widen, declines with age. The result is a microvascular network that is both structurally reduced and functionally impaired: fewer vessels, less responsive to the metabolic demands of exercise, and offering more resistance to the diffusion of oxygen at every step.
A Trainable Microvasculature
Crucially, none of this is fixed. Microvascular networks are among the more plastic structures in adult biology, and exercise is one of the most potent stimuli known to remodel them.
During sustained aerobic exercise, blood flow through working muscle increases dramatically, and that increase imposes a mechanical force called shear stress on the walls of blood vessels. Endothelial cells are exquisitely sensitive to shear stress: it activates signaling pathways that upregulate the production of nitric oxide and pro-angiogenic factors including vascular endothelial growth factor (VEGF). Over weeks and months of repeated training, these signals drive capillary proliferation, the sprouting of new capillary branches into the muscle tissue. Diffusion distances shorten. Endothelial function improves. The peripheral resistance to oxygen falls. [2]
In parallel, the same exercise stress that drives angiogenesis also activates PGC-1α and the mitochondrial biogenesis program, rebuilding the oxidative machinery that aging has eroded. The two adaptations are not independent; they are coordinated responses to the same metabolic challenge, and they reinforce each other. More capillaries mean more efficient oxygen delivery to more mitochondria. More mitochondria mean a greater capacity to consume the oxygen that improved capillary density delivers.
But not all exercise applies the same stress, and not all training produces the same pattern of remodeling. Different modalities, varying in intensity, duration, and the degree to which they recruit different muscle fiber types, engage these biological pathways to different degrees [2]. A 2024 comprehensive review and meta-regression by Molmen and colleagues compared the effects of endurance training, high-intensity interval training, and sprint interval training on mitochondrial content, capillary remodeling, and VO₂ max [3]. Its findings offer a practical framework for a question that follows directly from everything discussed so far: if the goal is to rebuild peripheral oxygen extraction capacity in aging muscle, what kind of training gives the best return on effort?
The two adaptations are not independent; they are coordinated responses to the same metabolic challenge, and they reinforce each other. More capillaries mean more efficient oxygen delivery to more mitochondria. More mitochondria mean a greater capacity to consume the oxygen that improved capillary density delivers.
Endurance Training: The Most Reliable Builder of Capillary Networks
Traditional endurance training, variously described as moderate continuous training, long slow distance, or more recently Zone 2, consists of sustained effort at relatively low to moderate intensity. In practice, this typically means sessions lasting 60 minutes or more at intensities at or below 60 to 65% of VO₂ max [3,4], an effort level at which a person can still hold a conversation but is working steadily and continuously. Familiar examples include brisk cycling at a pace that feels sustainable but not easy, steady incline walking for 30 to 60 minutes, or continuous rowing at moderate effort.
Of all the training modalities examined by Molmen and colleagues, endurance training proved the most consistent and reliable driver of both mitochondrial and vascular remodeling [3]. The biological logic is straightforward. Because endurance exercise sustains elevated blood flow through working muscle for extended periods, it exposes the walls of blood vessels to prolonged shear stress, the mechanical signal that most potently drives angiogenic signaling and capillary proliferation. At the same time, the prolonged reliance on oxidative metabolism required to sustain continuous effort repeatedly activates PGC-1α and the mitochondrial biogenesis program. The muscles are metabolically stressed for long enough that both sets of adaptations are robustly engaged.
The quantitative results from the meta-regression reflect this. Across the studies synthesized, endurance training produced a roughly 22.7% increase in mitochondrial content over 8 to 10 weeks, alongside a 15.0% increase in the capillary-to-fiber ratio, a 13.3% increase in capillary density, and a 12.5% improvement in VO₂ max [3].
Perhaps equally important is the time course of these adaptations. Mitochondrial remodeling begins rapidly: approximately 13.7% of the total mitochondrial increase observed over a full training block occurs within the first two weeks alone [3]. The body responds quickly to the metabolic demand. Capillary remodeling follows a similar early trajectory but appears to plateau after approximately four weeks [3], suggesting that the initial structural gains in the microvasculature are relatively accessible, but that sustaining further vascular growth likely requires progressive increases in training volume, intensity, or some combination of both.
This temporal pattern carries a practical implication. The peripheral adaptations most relevant to oxygen extraction, the ones that aging progressively erodes, begin accumulating within days of starting a consistent endurance program. The barrier to entry, biologically speaking, is lower than many people assume.
The quantitative results from the meta-regression reflect this. Across the studies synthesized, endurance training produced a roughly 22.7% increase in mitochondrial content over 8 to 10 weeks, alongside a 15.0% increase in the capillary-to-fiber ratio, a 13.3% increase in capillary density, and a 12.5% improvement in VO₂ max
HIIT: Time-Efficient Mitochondrial Remodeling with Strong VO₂ Max Gains
Where endurance training works by sustaining metabolic demand over long periods, high-intensity interval training (HIIT) applies a fundamentally different stimulus: repeated short bouts of near-maximal aerobic effort, typically at or above 87% of VO₂ max, separated by recovery periods that allow partial but not complete restoration [29]. In practice, this might look like four four-minute intervals at an intensity that makes conversation difficult, separated by three minutes of easy effort, or ten one-minute hard efforts alternated with one minute easy. The specific structure varies, but the physiological logic is consistent: drive oxygen demand and metabolic stress as high as possible, recover just enough to repeat it, and do so several times within a single session.
That repeated metabolic contrast, high oxygen demand, rapidly accumulating lactate, and intense energy turnover, create a particularly potent signal for mitochondrial remodeling. The sharp swings between high demand and partial recovery appear to activate PGC-1α and related signaling pathways more acutely than steady-state exercise at lower intensities, even when the total duration of effort is shorter.
The results from the Molmen meta-regression reflect this. Over approximately ten weeks, HIIT produced a 27.0% increase in mitochondrial content, a 13.3% increase in the capillary-to-fiber ratio, a 6.8% increase in capillary density, and a 12.1% improvement in VO₂ max [3]. The mitochondrial gains are notably larger than those seen with endurance training, and when adjusted for the time actually spent training, HIIT was estimated to be approximately 1.7 times more time-efficient for driving mitochondrial adaptations [3].
The capillary density numbers, however, tell a more nuanced story. Although the capillary-to-fiber ratio improved comparably to endurance training, the increase in capillary density measured as capillaries per square millimeter of muscle cross-section was more modest. One plausible explanation is that HIIT tends to increase muscle fiber size more than steady endurance work does. When fibers grow larger, the same number of capillaries is now serving a greater tissue volume, which can dilute the apparent density even if the total number of capillaries has increased. It is a geometric artifact of hypertrophy rather than evidence of inferior vascular adaptation.
As with endurance training, the angiogenic gains from HIIT appear to plateau around the four-week mark [3], reinforcing the pattern seen across modalities: the microvascular network responds early and then requires a new or intensified stimulus to keep remodeling.
For older adults in particular, HIIT represents a compelling option when training time is limited and the priority is rebuilding mitochondrial capacity quickly. Its vascular benefits are real, if somewhat smaller than those produced by endurance training, and its VO₂ max gains are comparable. The tradeoff is recovery demand, a consideration that becomes increasingly relevant with age and one we will return to.
The mitochondrial gains are notably larger than those seen with endurance training, and when adjusted for the time actually spent training, HIIT was estimated to be approximately 1.7 times more time-efficient for driving mitochondrial adaptations
Sprint Interval Training: The Fastest Mitochondrial Signal per Minute
Sprint interval training takes the logic of HIIT to its extreme. Where HIIT operates at near-maximal aerobic intensity, sprint interval training (SIT) consists of true all-out efforts, supramaximal bursts lasting 30 seconds or less, followed by recovery periods that are deliberately long relative to the work interval, typically two to four times the duration of the sprint itself [3,5,6]. In practice this might look like four to six maximal efforts on a stationary bike, each lasting 20 to 30 seconds, with two to four minutes of easy pedaling between each. Or six short hill sprints with a full walk-down recovery. The total time spent at high intensity in a single SIT session can be as little as two to three minutes.
What makes SIT physiologically interesting is the nature of the stress it creates in that brief window. An all-out sprint lasting 20 to 30 seconds depletes ATP at an extraordinary rate, drives lactate to very high levels almost immediately, and recruits a large fraction of the available muscle mass simultaneously. This acute metabolic crisis, despite its brevity, appears to be a remarkably potent trigger for mitochondrial biogenesis signaling. The cell responds to the severity of the stress, not just its duration.
The numbers from the Molmen meta-regression bear this out. SIT produced a 27.0% increase in mitochondrial content, comparable to HIIT and substantially larger than endurance training, along with a 10.4% increase in the capillary-to-fiber ratio [3]. Notably, approximately 21.5% of the total mitochondrial adaptation occurred within the first two weeks, an even faster early response than that seen with the other modalities. VO₂ max improvements ranged from 6.6 to 8.9%.
SIT's most striking attribute, however, is its time efficiency. When mitochondrial and VO₂ max gains are normalized to total training time, SIT generated approximately three to five times greater VO₂ max improvement per hour of exercise than either endurance training or HIIT [3]. For a modality that can be completed in under fifteen minutes including recovery, that ratio is remarkable.
The limitations are real, though, and they matter particularly in the context of aging. SIT's vascular remodeling effects are its weakest dimension. Capillary density showed no significant average increase across the studies examined, and the capillary-to-fiber ratio gains were more modest than those produced by endurance training [3]. The mitochondrial improvements also appear to plateau earlier than with other modalities, suggesting that SIT delivers a rapid initial stimulus but may not sustain progressive adaptation over longer training periods in the way that endurance exercise does.
The picture that emerges is of a modality with a very specific profile: exceptional for rapidly rebuilding mitochondrial capacity, particularly in time-constrained individuals, but insufficient on its own as a strategy for the sustained capillary expansion that peripheral oxygen extraction ultimately depends on. In the context of aerobic aging, where both mitochondrial loss and capillary rarefaction are contributing to rising peripheral resistance, SIT addresses one problem more effectively than the other.
What About Resistance Training?
Resistance training occupies its own category in this discussion, because it targets a fundamentally different biological problem. Where endurance and interval training expand the body's capacity to deliver and consume oxygen, resistance training primarily expands force production capacity, preserving the muscle mass, strength, and structural integrity that underpin physical independence and survival in older age. These are not the same adaptation, and the distinction matters.
At the mechanistic level, resistance training produces hypertrophy, the growth of individual muscle fibers, along with a degree of microvascular remodeling. What it does not reliably produce, at least in young healthy individuals, is robust mitochondrial biogenesis [6]. Studies show that resistance training performed to volitional failure increases the cross-sectional area of both Type I and Type II muscle fibers, and alongside that growth, capillary-to-fiber ratio, capillary contacts, and capillary exchange capacity all tend to improve [6,7,8]. Capillary density, however, measured as capillaries per unit area of muscle cross-section, often remains unchanged.
This pattern reveals something important about how resistance training remodels the microvasculature. The capillary network appears to expand roughly in proportion to fiber growth, preserving the geometric relationship between vessels and the tissue they supply as the muscle grows larger, rather than adding capillaries beyond what fiber size alone would require. It is maintenance of diffusion capacity rather than expansion of it, a meaningful distinction from the absolute angiogenic gains produced by sustained endurance exercise.
The mitochondrial story is similarly nuanced. As muscle fibers grow through resistance training, their volume increases faster than the number of mitochondria within them, producing what researchers describe as mitochondrial dilution: a lower mitochondrial density within a given fiber even if the total number of mitochondria is preserved or slightly increased [6,7,8]. On the surface, this sounds like a disadvantage, but the functional implications are more complex. Several studies report preserved or even improved mitochondrial respiratory efficiency and electron transport chain activity following resistance training, suggesting that the mitochondria that remain are functioning well despite being more spread out within a larger fiber [6,7,8].
Age and baseline fitness also appear to modify this picture considerably. Older or less active individuals tend to show more consistent mitochondrial functional improvements from resistance training than their younger or fitter counterparts [6,7,8], possibly because they are starting from a lower baseline of both muscle mass and mitochondrial quality, leaving more room for adaptation on both dimensions simultaneously.
Taken together, resistance training is indispensable for healthy aging, but for reasons that sit somewhat apart from the peripheral oxygen extraction story this article has been building. It preserves the muscle mass that sarcopenia progressively erodes, maintains the structural scaffolding within which capillaries and mitochondria must operate, and supports microvascular supply in proportion to fiber size. What it does not do, at least not reliably, is drive the robust mitochondrial biogenesis and capillary proliferation that aerobic training produces. It should be understood as a complementary pillar of any training approach for aging, essential but not interchangeable with the modalities that most directly target the peripheral resistance at the heart of age-related VO₂ max decline.
Putting It All Together: Choosing the Right Tool for the Job
From a longevity perspective, these training modalities should not be framed as competitors. They are different tools that stress different biological systems:
- Endurance training = best for building capillary networks and aerobic foundation
- HIIT = best for time-efficient VO₂max + mitochondrial remodeling
- SIT = best for rapid mitochondrial signaling per minute trained
- Resistance training = best for strength, muscle preservation, and aging resilience
For healthspan, the optimal strategy is rarely specialization. Instead, the strongest approach is training diversity: combining endurance and intervals to support vascular–mitochondrial infrastructure while using resistance training to preserve muscle mass, power, and independence.
Practical Translation for Older Adults and Time-Constrained Individuals
Understanding the biology of aerobic aging is one thing. Translating it into a training practice that a real person can sustain across decades is another challenge entirely. Aging trajectories vary widely, and the practical constraints of real life often become more limiting than physiology itself. Time scarcity, orthopedic limitations, declining recovery capacity, and reduced exercise tolerance all shape what training is actually feasible, particularly for older adults [39,40]. For these populations, the goal is not to identify the single best modality and pursue it to its theoretical maximum. It is to build a program that is biologically effective, adaptable to circumstance, and sustainable across years.
Here the emerging evidence becomes genuinely encouraging. The data collectively challenge the assumption that large training volumes are required to produce meaningful mitochondrial and vascular adaptation. Mitochondrial content and oxidative capacity can respond rapidly, particularly when intensity is applied strategically, while capillary remodeling often occurs early in a training block and does not scale linearly with ever-increasing volume. More training is not always proportionally better. For many individuals, especially those with limited time or limited tolerance, the most important step is identifying the minimum stimulus that reliably produces adaptation, then repeating it consistently enough for those benefits to accumulate over time.
A practical principle follows from this. Intensity can often substitute for time, but only when applied thoughtfully. Higher-intensity interval approaches can deliver greater physiological return per unit of training time, making them particularly appealing for time-constrained individuals. Yet the same intensity that makes interval training efficient also increases injury risk, recovery demand, and discomfort, considerations that become progressively more significant with age. The optimal strategy is therefore not maximal intensity, but appropriate intensity: enough to activate mitochondrial and cardiovascular remodeling without exceeding the individual's orthopedic or recovery limits.
Rather than viewing endurance, interval, sprint, and resistance training as competing options, the evidence supports a complementary framework in which each modality contributes something distinct. Short blocks of high-intensity training can rapidly stimulate mitochondrial adaptation, particularly after sedentary periods, but tend to plateau early. Endurance and HIIT provide more gradual, sustainable remodeling, particularly of the microvasculature that supports long-term oxygen delivery. Resistance training, while not a primary driver of mitochondrial biogenesis, remains essential for preserving the muscle mass, strength, and functional independence within which all of these aerobic adaptations must operate.
In practice, this opens the door to a flexible sequencing approach that fits real life. A time-limited individual might use brief interval sessions during a demanding month, then shift toward longer steady-state training when schedule and recovery allow. An older adult managing joint health might prioritize low-impact modalities such as cycling, incline walking, or rowing that provide a meaningful cardiovascular stimulus without excessive orthopedic stress, while keeping resistance training as the anchor for strength and mobility. Rotating training styles across weeks or seasons allows the body to tap different adaptive pathways, avoid plateaus, reduce overuse injury risk, and sustain engagement. For healthspan, where the goal is not a short-term performance peak but the preservation of mitochondrial and vascular function across the lifespan, that kind of long-term consistency matters more than any particular session.
Minimum Effective Dose: Practical Weekly Templates
One of the most encouraging findings in modern exercise physiology is that meaningful mitochondrial and vascular benefits do not require extreme training volumes. The body responds strongly to relatively modest doses, especially when intensity is applied strategically. The templates below offer evidence-informed starting points that balance time efficiency with long-term sustainability. They are not rigid prescriptions, but practical frameworks that can be adjusted based on age, injury history, baseline fitness, and recovery capacity.
The Busy Week Minimum (approximately 30 minutes per week)
Goal: Preserve mitochondrial signaling and aerobic capacity when time is severely limited. Best for high-workload periods, travel, or maintenance mode.
The simplest version of this template involves two short HIIT sessions per week, each lasting 10 to 15 minutes, built around six one-minute hard efforts alternated with one minute easy, plus a brief warm-up and cool-down. For those who prefer an even more compressed stimulus, a single SIT session per week, consisting of four to six 20 to 30 second all-out efforts with long recovery intervals, can fit within the same time window.
A small number of intense bouts is sufficient to reactivate mitochondrial biogenesis signaling and preserve VO₂ max surprisingly well, particularly in individuals who have trained previously. Think of this as the minimum dose required to keep the system online during periods when more is not possible.
The High-Return Healthspan Dose (approximately 60 to 90 minutes per week)
Goal: Improve VO₂ max, mitochondrial content, and sustain vascular remodeling. Best for most people pursuing long-term healthspan.
A practical weekly structure at this dose might include one HIIT session of 15 to 25 minutes, built around four four-minute hard intervals with three minutes of easy recovery between each, combined with one endurance session of 30 to 45 minutes at a conversational Zone 2 pace. An optional third session of easy walking or light cycling adds low-stress blood flow and recovery stimulus without meaningful additional fatigue.
This combination sits in a genuine sweet spot. The interval session drives robust mitochondrial adaptation while the sustained endurance work provides enough prolonged blood flow to support capillary remodeling and vascular health. Together, they address both sides of the peripheral limitation that this article has been examining, in under 90 minutes per week.
The Long-Term Longevity Builder (approximately 2.5 to 3 hours per week)
Goal: Maximize mitochondrial capacity and build long-term vascular durability. Best for individuals prioritizing deep aerobic reserve and metabolic resilience.
At this investment level, a weekly structure might include two endurance sessions of 40 to 60 minutes each, one HIIT session of 20 to 30 minutes of total work, and an optional easy recovery session. This volume tends to produce the most reliable long-term improvements in capillary density, mitochondrial remodeling, and aerobic durability. Critically, it builds a deeper and more diverse physiological reserve, which becomes increasingly valuable as the decades accumulate and the margin between functional capacity and functional threshold narrows.
Where Resistance Training Fits
For healthspan, aerobic training addresses only half of the biological problem that aging presents. Preserving independence and reducing frailty risk requires maintaining the muscle mass, strength, and power that resistance training uniquely provides. The minimum effective dose for most people is two sessions per week of 30 to 60 minutes, focused on the fundamental movement patterns: squat, hinge, push, pull, and carry.
Resistance training does not compete with the aerobic templates above. It completes them. The mitochondrial and angiogenic benefits of aerobic exercise operate within skeletal muscle, and that muscle must be preserved to serve as the substrate for those adaptations. Without it, the oxygen cascade has progressively less tissue to supply and less machinery to run.
The Long Game
The biology described in this article unfolds slowly. VO₂ max does not collapse overnight. Mitochondria do not vanish in a season. Capillary networks do not thin across a year. The oxygen cascade deteriorates the way most consequential biological processes do: gradually, simultaneously, across multiple systems, over decades, in ways that are nearly invisible until they are not.
That time horizon is both the challenge and the opportunity.
The challenge is that the consequences of inactivity accumulate long before they become legible. A person in their late forties who is sedentary but functional has no immediate signal that their mitochondrial density is declining, that their capillary networks are thinning, or that the peripheral side of their Fick equation is quietly accounting for a growing fraction of their aerobic limitation. The biology is patient. It does not announce itself.
The opportunity is that the adaptations work on the same time horizon. Mitochondrial biogenesis begins within days of a meaningful training stimulus. Capillary remodeling takes hold within weeks. VO₂ max responds within months. And crucially, the biological machinery responsible for these adaptations remains responsive well into the later decades of life, more responsive, the evidence suggests, than most people assume. The system that aging slowly erodes is the same system that exercise can partially rebuild. Not indefinitely, and not completely. But meaningfully, and for longer than the conventional narrative about aging has typically allowed.
What this study adds to that picture is precision. For the first time with this level of quantitative resolution, we can see that the age-related decline in VO₂ max is not a single-system failure. It is a coordinated deterioration across the entire oxygen transport chain, from the cardiac muscle that pumps blood, to the capillaries that deliver it, to the mitochondria that consume it. By late middle age, nearly half of the total limitation on aerobic capacity is peripheral in origin. The heart is still part of the story. But the muscles, and everything inside them, are nearly an equal part.
That finding reframes the question that exercise physiology has been asking for over a century. The question was never really whether VO₂ max declines. It was always why, and therefore what to do about it. The answer, it turns out, is not to focus narrowly on the heart or narrowly on the muscle, but to train the full length of the oxygen cascade, consistently, across the decades during which its slow remodeling is underway.
The decisions made in the fourth and fifth decades of life are, in a meaningful biological sense, shaping the physiological ceiling of the seventh and eighth. Not because any single workout matters that much, but because the cumulative signal of thousands of workouts, across years and decades, is what determines whether the oxygen cascade stays permeable or progressively closes down.
That is the long game. And the biology suggests it is very much worth playing.
- Capelli, C., Ferretti, G., di Prampero, P.E. et al. Cardiovascular and peripheral factors affecting the decay of maximal oxygen uptake across the spectrum of age in humans. Eur J Appl Physiol (2025). https://doi.org/10.1007/s00421-025-06031-6
- Spanoudaki M, Giaginis C, Karafyllaki D, Papadopoulos K, Solovos E, Antasouras G, Sfikas G, Papadopoulos AN, Papadopoulou SK. Exercise as a Promising Agent against Cancer: Evaluating Its Anti-Cancer Molecular Mechanisms. Cancers (Basel). 2023 Oct 25;15(21):5135. doi: 10.3390/cancers15215135. PMID: 37958310; PMCID: PMC10648074.
- Kraus, W. E., Powell, K. E., Haskell, W. L., Janz, K. F., Campbell, W. W., Jakicic, J. M., Troiano, R. P., Sprow, K., Torres, A., & Piercy, K. L. (2019). Physical activity, all‑cause and cardiovascular mortality, and cardiovascular disease. Medicine & Science in Sports & Exercise, 51(6), 1270‑1281. https://doi.org/10.1249/MSS.0000000000001939
- Tomás, M. T., Galán‑Mercant, A., Alvarez Carnero, E., & Fernandes, B. (2017). Functional capacity and levels of physical activity in aging: A 3‑year follow‑up. Frontiers in Medicine, 4, 244. https://doi.org/10.3389/fmed.2017.00244
- Brogno, B. (2025). Aging with strength: Functional training to support independence and quality of life. Inquiry, 62, 00469580251348133. https://doi.org/10.1177/00469580251348133
- Hyvärinen, M., Kankaanpää, A., Rantalainen, T., Rantanen, T., Laakkonen, E. K., & Karavirta, L. (2025). Body composition and functional capacity as determinants of physical activity in middle‑aged and older adults: A cross‑sectional analysis. European Review of Aging and Physical Activity, 22, Article 6. https://doi.org/10.1186/s11556‑025‑00372‑z
- Satsangi, M., & Tadi, P. (2023). Physiology, vascular. In StatPearls [Internet]. StatPearls Publishing. Retrieved from the National Center for Biotechnology Information Bookshelf
- Cooper, G. M., & Hausman, R. E. (2000). Mitochondria. In The Cell: A Molecular Approach (2nd ed.). Sinauer Associates. (NCBI Bookshelf entry
- Ungvari, Z., Sonntag, W. E., & Csiszar, A. (2010). Mitochondria and aging in the vascular system. Journal of Molecular Medicine, 88(10), 1021‑1027. https://doi.org/10.1007/s00109‑010‑0667‑5
- Wang, S., Pi, Y., Yang, X., et al. (2023). New insights into vascular aging: Emerging role of mitochondria function. Biomedicine & Pharmacotherapy, 162, Article 114592. https://doi.org/10.1016/j.biopha.2023.114592
- Jani, B., & Rajkumar, C. (2006). Ageing and vascular ageing. Postgraduate Medical Journal, 82(968), 357‑362. https://doi.org/10.1136/pgmj.2005.036053
- Strasser, B., & Burtscher, M. (2018). Survival of the fittest: VO₂max, a key predictor of longevity? Frontiers in Bioscience (Landmark Edition), 23, 1505‑1516. https://doi.org/10.2741/4657
- Pette, D., & Staron, R. S. (2000). Myosin isoforms, muscle fiber types, and transitions. Microscopy Research and Technique, 50(6), 500‑509. https://doi.org/10.1002/1097‑0029(20000915)50:6
- Plotkin, D. L., Roberts, M. D., Haun, C. T., & Schoenfeld, B. J. (2021). Muscle fiber type transitions with exercise training: Shifting perspectives. Sports (Basel), 9(9), 127. https://doi.org/10.3390/sports9090127
- Poole, D. C., Copp, S. W., Ferguson, S. K., & Musch, T. I. (2012). Anatomy of skeletal muscle and its vascular supply. In Skeletal Muscle Circulation (Chapter 2). Morgan & Claypool Life Sciences
- Ørtenblad, N., Nielsen, J., Boushel, R., Saltin, B., & Holmberg, H.‑C. (2018). The muscle fiber profiles, mitochondrial content, and enzyme activities of the exceptionally well‑trained arm and leg muscles of elite cross‑country skiers. Frontiers in Physiology, 9, 1031. https://doi.org/10.3389/fphys.2018.01031
- Hickson, R. C., Hidaka, K., & Foster, C. (1994). Skeletal muscle fiber type, resistance training, and strength‑related performance. Medicine & Science in Sports & Exercise, 26(5), 593‑598
- Memme, J. M., Erlich, A. T., Phukan, G., & Hood, D. A. (2021). Exercise and mitochondrial health. The Journal of Physiology, 599(3), 803‑817. https://doi.org/10.1113/JP278853
- Musci, R. V., Hamilton, K. L., & Linden, M. A. (2019). Exercise‑induced mitohormesis for the maintenance of skeletal muscle and healthspan extension. Sports, 7(7), Article 170. https://doi.org/10.3390/sports7070170
- Moore, T. M., et al. (2019). The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Molecular Metabolism, 21, 51‑67. https://doi.org/10.1016/j.molmet.2018.11.012
- Halling, J. F., & Pilegaard, H. (2020). PGC‑1α‑mediated regulation of mitochondrial function and physiological implications. Applied Physiology, Nutrition, and Metabolism, 45(9), 927‑936. https://doi.org/10.1139/apnm‑2020‑0005
- Geng, T., Li, P., Okutsu, M., Yin, X., Kwek, J. Y., Zhang, M., & Yan, Z. (2010). PGC‑1α plays a functional role in exercise‑induced mitochondrial biogenesis and angiogenesis but not fiber‑type transformation in mouse skeletal muscle. American Journal of Physiology – Cell Physiology, 298(3), C??–C?? (issue pages). https://doi.org/10.1152/ajpcell.00481.2009
- Chen, L., Qin, Y., Liu, B., Gao, M., Cai, Y., et al. (2022). PGC‑1α‑mediated mitochondrial quality control: Molecular mechanisms and implications for heart failure. Frontiers in Cell and Developmental Biology, 10, Article 871357. https://doi.org/10.3389/fcell.2022.871357
- Ross, M., Kargl, C. K., Ferguson, R. G., Poggi, T. P., & Vila, J. (2023). Exercise‑induced skeletal muscle angiogenesis: Impact of age, sex, angiocrines and cellular mediators. European Journal of Applied Physiology, 123(7), 1415‑1432. https://doi.org/10.1007/s00421‑022‑05128‑6
- Duscha, B. D., Kraus, W. E., Keteyian, S. J., Sullivan, M. J., Green, H. J., Schachat, F. H., Pippen, A. M., Brawner, C. A., Blank, J. M., & Annex, B. H. (1999). Capillary density of skeletal muscle: A contributing mechanism for exercise intolerance in class II–III chronic heart failure independent of other peripheral alterations. Journal of the American College of Cardiology, 33(7), 1956‑1963. https://doi.org/10.1016/S0735‑1097(99)00101‑1
- Arany, Z., Foo, S.‑Y., Ma, Y., Ruas, J. L., Bommi‑Reddy, A., Girnun, G., Cooper, M., Laznik, D., Chinsomboon, J., Rangwala, S. M., Baek, K. H., Rosenzweig, A., & Spiegelman, B. M. (2008). HIF‑independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC‑1α. Nature, 451, 1008‑1012. https://doi.org/10.1038/nature06613
- Løck, L., Hellsten, Y., Fentz, J., Lyngby, S. S., Wojtaszewski, J. F. P., Hidalgo, J., & Pilegaard, H. (2009). PGC‑1α mediates exercise‑induced skeletal muscle VEGF expression in mice. American Journal of Physiology – Endocrinology and Metabolism, 297(1), E??–E?? (issue pages). https://doi.org/10.1152/ajpendo.00076.2009
- Burnham, R., Martin, T., Stein, R., Bell, G., MacLean, I., & Stewart, C. (1997). Skeletal muscle fibre type transformation following spinal cord injury. Spinal Cord, 35(2), 86‑91. https://doi.org/10.1038/sj.sc.3100364
- Mølmen, K. S., Almquist, N. W., & Skattebo, Ø. (2025). Effects of exercise training on mitochondrial and capillary growth in human skeletal muscle: A systematic review and meta‑regression. Sports Medicine, 55(1), 115‑144. https://doi.org/10.1007/s40279‑024‑02120‑2,
- MacIntosh, B. R., Murias, J. M., Keir, D. A., & Weir, J. M. (2021). What is moderate to vigorous exercise intensity? Frontiers in Physiology, 12, Article 682233. https://doi.org/10.3389/fphys.2021.682233
- Ingjer, F. (1979). Effects of endurance training on muscle fibre ATP‑ase activity, capillary supply and mitochondrial content in man. The Journal of Physiology, 294, 419‑432. https://doi.org/10.1113/jphysiol.1979.sp012938
- Baum, O., Gubeli, J., Friese, S., Tschanz, E., Malik, C., Odriozola, A., Graber, F., & Hoppeler, H. (2015). Angiogenesis‑related ultrastructural changes to capillaries in human skeletal muscle in response to endurance exercise. Journal of Applied Physiology, 119(10), 1119‑1127. https://doi.org/10.1152/japplphysiol.00594.2015
- Milanović, Z., Sporiš, G., & Weston, M. (2015). Effectiveness of high‑intensity interval training (HIT) and continuous endurance training for VO₂max improvements: A systematic review and meta‑analysis of controlled trials. Sports Medicine, 45, 1469‑1481. https://doi.org/10.1007/s40279‑015‑0365‑0
- Jacobs, R. A., Flück, D., Bonne, T. C., Bürgi, S., Christensen, P. M., & Lundby, C. (2013). Improvements in exercise performance with high‑intensity interval training coincide with an increase in skeletal muscle mitochondrial content and function. Journal of Applied Physiology, 115(6), 785‑793. https://doi.org/10.1152/japplphysiol.00445.2013
- Mitchell, E. A., Martin, N. R. W., Turner, M. C., Taylor, C. W., & Ferguson, R. A. (2018). The combined effect of sprint interval training and post‑exercise blood flow restriction on critical power, capillary growth, and mitochondrial proteins in trained cyclists. Journal of Applied Physiology, 126(1), 79‑90. https://doi.org/10.1152/japplphysiol.01082.2017
- Parry, H. A., Roberts, M. D., & Kavazis, A. N. (2020). Human skeletal muscle mitochondrial adaptations following resistance exercise training. International Journal of Sports Medicine, 41(6), 349‑359. https://doi.org/10.1055/a‑1121‑7851
- Porter, C., Reidy, P. T., Bhattarai, N., Sidiropoulos, N., & Rasmussen, B. B. (2015). Resistance exercise training alters mitochondrial function in human skeletal muscle. Medicine & Science in Sports & Exercise, 47(9), 1922‑1931. https://doi.org/10.1249/MSS.0000000000000605
- Holloway, T. N., Morton, R. W., Oikawa, S. Y., McKellar, S., Baker, S. K., & Phillips, S. M. (2018). Microvascular adaptations to resistance training are independent of load in resistance‑trained young men. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology, 315(2), R293‑R301. https://doi.org/10.1152/ajpregu.00118.2018
- Hyvärinen, M., Kankaanpää, A., Rantalainen, T., Rantanen, T., Laakkonen, E. K., & Karavirta, L. (2025). Body composition and functional capacity as determinants of physical activity in middle‑aged and older adults: A cross‑sectional analysis. European Review of Aging and Physical Activity, 22, Article 6. https://doi.org/10.1186/s11556‑025‑00372‑z
- Balicki, P., Sołtysik, B. K., Borowiak, E., Kostka, T., & Kostka, J. (2025). Activities of daily living limitations in relation to the presence of pain in community‑dwelling older adults. Scientific Reports, 15, Article 15027. https://doi.org/10.1038/s41598‑025‑00241‑w
Related studies